Chapter 2 Selbal: selection of balances
Selbal is a forward selection algorithm for the identification of two groups of variables whose balance is most associated with the response variable (Rivera-Pinto et al. 2018). Selbal R package is available on GitHub (https://github.com/UVic-omics/selbal) and can be installed with devtools:
for non-Windows users:
devtools::install_github(repo = "UVic-omics/selbal")for Windows users:
devtools::install_url(url="https://github.com/UVic-omics/selbal/archive/master.zip",
INSTALL_opt= "--no-multiarch")For a detailed description of selbal see the vignette: https://htmlpreview.github.io/?https://github.com/UVic-omics/selbal/blob/master/vignettes/vignette.html
We generated a wrapper function called selbal_wrapper() that will help us to handle the output of selbal. The selbal_wrapper() function is uploaded via functions.R.
2.1 Crohn case study
For binary outcomes, selbal() requires that dependent variable Y is given as a factor and it implements logistic regression. If Y is numeric, selbal() implements linear regression.
The dependent variable in Crohn dataset is a factor:
class(y_Crohn)## [1] "factor"The performance measure (logit.acc) of the selected balance for binary outcomes is the AUC (default) or the proportion of explained deviance (Dev). For comparison with the other methods we will use Dev and will set the maximum number of variables (maxV) to be selected equal to 12 (maxV = 12).
selbal_Crohn <- selbal(x = x_Crohn, y = y_Crohn, maxV = 12,
logit.acc = 'Dev', draw = F)The output of selbal() is a list and we can get the different elements of the list by indexing.
To visualise the results of selbal, we recommend the new balance representation (global.plot2):
# dev.off() # clean plots window when you run in Console
grid.draw(selbal_Crohn$global.plot2) To improve the readability of codes and to compare more easily with the other two methods, we use selbal_wrapper() to handle the output from selbal():
Crohn.results_selbal <- selbal_wrapper(result = selbal_Crohn, X = x_Crohn) The number of selected variables:
Crohn.results_selbal$numVarSelect## [1] 12The names of selected variables:
Crohn.results_selbal$varSelect## [1] "g__Roseburia" "g__Eggerthella"
## [3] "g__Dialister" "g__Streptococcus"
## [5] "f__Peptostreptococcaceae_g__" "g__Bacteroides"
## [7] "g__Aggregatibacter" "g__Adlercreutzia"
## [9] "g__Dorea" "g__Oscillospira"
## [11] "o__Clostridiales_g__" "g__Blautia"For visualisation, we can use selbal_like_plot() which can also be used in other two methods (see Chapter 5).
Crohn.selbal_pos <- Crohn.results_selbal$posVarSelect
Crohn.selbal_neg <- Crohn.results_selbal$negVarSelect
selbal_like_plot(pos.names = Crohn.selbal_pos, neg.names = Crohn.selbal_neg,
Y = y_Crohn, selbal = TRUE,
FINAL.BAL = Crohn.results_selbal$finalBal)
2.2 HFHS-Day1 case study
The analysis on HFHSday1 data is similar to Crohn data.
First, we need to check if the dependent variable Y is a factor.
class(y_HFHSday1)## [1] "factor"We set the maximum number of variables to be selected equal to 2 (maxV = 2) according to tuning function selbal.cv() (not shown here) (Rivera-Pinto et al. 2018):
selbal_HFHSday1 <- selbal(x = x_HFHSday1, y = y_HFHSday1, maxV = 2,
logit.acc = 'Dev', draw = F)We then use selbal_wrapper() to handle the results:
HFHS.results_selbal <- selbal_wrapper(result = selbal_HFHSday1, X = x_HFHSday1) The number of selected variables:
HFHS.results_selbal$numVarSelect## [1] 2The names of selected variables:
HFHS.results_selbal$varSelect## [1] "290253" "263479"For visualisation, we then use selbal_like_plot().
HFHS.selbal_pos <- HFHS.results_selbal$posVarSelect
HFHS.selbal_neg <- HFHS.results_selbal$negVarSelect
selbal_like_plot(pos.names = HFHS.selbal_pos, neg.names = HFHS.selbal_neg,
Y = y_HFHSday1, selbal = TRUE,
FINAL.BAL = HFHS.results_selbal$finalBal,
OTU = T, taxa = taxonomy_HFHS)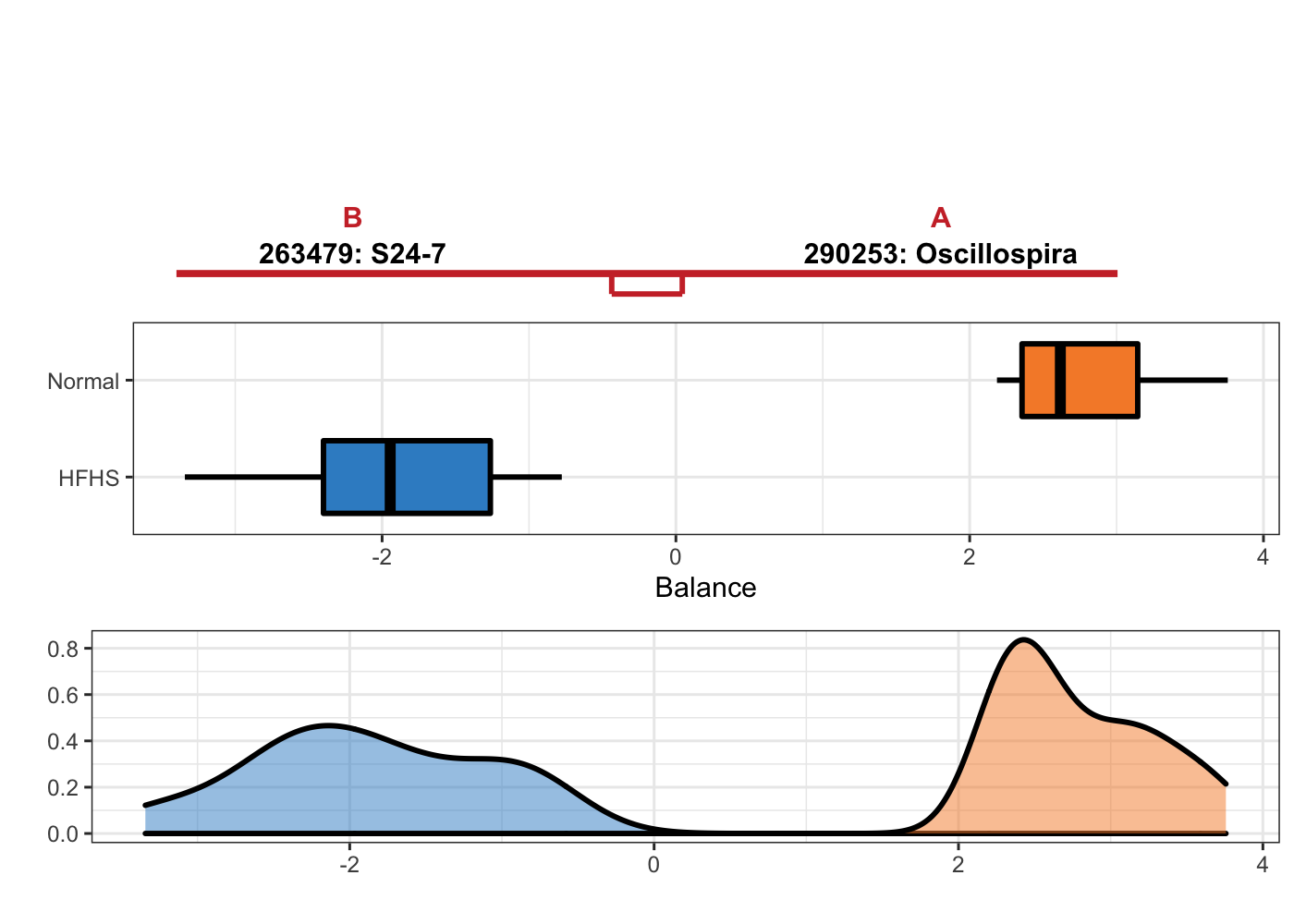
We also extract the taxonomic information of these selected OTUs.
HFHS.tax_selbal <- taxonomy_HFHS[which(rownames(taxonomy_HFHS) %in%
HFHS.results_selbal$varSelect), ]
kable(HFHS.tax_selbal[ ,2:6], booktabs = T)| Phylum | Class | Order | Family | Genus | |
|---|---|---|---|---|---|
| 290253 | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Oscillospira |
| 263479 | Bacteroidetes | Bacteroidia | Bacteroidales | S24-7 |
References
Rivera-Pinto, J, JJ Egozcue, Vera Pawlowsky-Glahn, Raul Paredes, Marc Noguera-Julian, and ML Calle. 2018. “Balances: A New Perspective for Microbiome Analysis.” MSystems 3 (4). Am Soc Microbiol: e00053–18.